
07022 Entwicklung von Software as a Medical Device – Medical Apps
Anforderungen der DIN EN 82304
|
Dieser Beitrag beschäftigt sich mit der regulatorischen Umsetzung von mehreren anzuwendenden Normen zur Entwicklung von Software as a Medical Device und explizit mit den Anforderungen für Medical Apps. Es wird klar abgegrenzt, wann eine Software als eigenständiges Medizinprodukt anzusehen ist. Diesbezüglich wird auf die Anforderungen und Zusammenhänge von IEC 62304 und IEC 82304-1 eingegangen.
Ziel ist eine Übersicht über die Herausforderungen und Abgrenzungen der unterschiedlichen regulatorischen Anforderungen in Bezug auf die Prozesse im Life-Cycle einer Medical App zu geben.
Wenn in diesem Beitrag von der DIN EN 62304 gesprochen wird, ist damit die DIN EN 62304:2006/AMD1:2015 gemeint, bei der DIN EN 82304-1 handelt es sich um die DIN EN 82304-1:2017. von: |
1 Definition und Abgrenzung
Bei jedem Produkt ist es fundamental wichtig, dass der Inverkehrbringer sich mit den zutreffenden regulatorischen Anforderungen auseinandersetzt. Dabei stellen sich für Inverkehrbringer bei einem Produkt wie z. B. einer App, die einen Zusammenhang mit medizinischen Daten besitzt, oftmals die Frage: Ist die App ein Medizinprodukt? Weiterhin gibt es eine Vielzahl von Bezeichnungen und Unterscheidungen für diese Produkte. In diesem Beitrag wird „Produkt” synonym für eine potenzielle Medical App, Software as a Medical Device, Medizinprodukt oder auch Nicht-Medizinprodukt verwendet. Man könnte auch die Bezeichnung „Betrachtungsgegenstand” wählen.
Einteilung entscheidend
Das Verständnis für die Unterschiede und die Einteilung der Produkte ist abhängig von der Zweckbestimmung und stellt die Grundlage für die anzuwendenden regulatorischen Anforderungen dar. Für den Inverkehrbringer hängt sehr viel von dieser Entscheidung ab, daher werden in diesem Beitrag die Unterschiede kurz erläutert. Die Haftungsrisiken von Medical Apps und die detaillierten Unterschiede der differenzierten Begriffe in Bezug auf Medizinproduktesoftware sind in dem Beitrag Medizinische Apps: Haftungsrisiken bereits ausführlich beschrieben (s. Kap. 04010).
Das Verständnis für die Unterschiede und die Einteilung der Produkte ist abhängig von der Zweckbestimmung und stellt die Grundlage für die anzuwendenden regulatorischen Anforderungen dar. Für den Inverkehrbringer hängt sehr viel von dieser Entscheidung ab, daher werden in diesem Beitrag die Unterschiede kurz erläutert. Die Haftungsrisiken von Medical Apps und die detaillierten Unterschiede der differenzierten Begriffe in Bezug auf Medizinproduktesoftware sind in dem Beitrag Medizinische Apps: Haftungsrisiken bereits ausführlich beschrieben (s. Kap. 04010).
Die wichtigste Fragestellung lautet zunächst immer: „Handelt es sich bei meinem Produkt um ein Medizinprodukt oder nicht?”
1.1 Ist es ein Medizinprodukt?
In der Praxis ist die Entscheidung, ob es sich um ein Medizinprodukt handelt, oftmals unklar. Davon sind Faktoren abhängig, wie zum Beispiel, ob der Einsatz des Produkts von der Krankenkasse bezahlt wird, ob das Produkt einen Zulassungsprozess durchlaufen muss oder ob das Produkt als DIGA (Digitale Gesundheitsanwendung) gelistet werden kann.
1.1.1 Definition MDR
In Europa lautet die Definition eines Medizinprodukts gemäß Artikel 2 Abs. 1 Medical Device Regulation (MDR) 2017/745:
MDR Artikel 2.1
„’Medizinprodukt’ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
„’Medizinprodukt’ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
| • | Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten, |
| • | Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen, |
| • | Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands, |
| • | Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper – auch aus Organ-, Blut- und Gewebespenden – stammenden Proben |
und dessen bestimmungsgemäße Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
| • | Produkte zur Empfängnisverhütung oder -förderung, |
| • | Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Artikel 1 Absatz 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.” |
Somit ist gemäß der Definition Medizinprodukt in der MDR klar, dass die in diesem Beitrag betrachteten Produkte Medizinprodukte sein können. Da diese Begriffsbestimmung noch immer nicht spezifisch genug ist, gibt es dazu unterschiedliche Erläuterungen.
1.1.2 Definition IMDRF
Zunächst betrachten wir die Ausführungen gemäß IMDRF (International Medical Device Regulators Forum) Software as a Medical Device (SaMD): Key Definitions vom Dezember 2013.
Hier werden zusätzliche Überlegungen zu der Definition Medizinprodukt aufgeführt.
| IMDRF ”5.2.3 Additional considerations for SaMDSaMD may also: |
| • | provide means and suggestions for mitigation of a disease; |
| • | provide information for determining compatibility, detecting, diagnosing, monitoring or treating physiological conditions, states of health, illnesses or congenital deformities; |
| • | be an aid to diagnosis, screening, monitoring, determination of predisposition; prognosis, prediction, determination of physiological status." |
Erweiterte Definition
Dies erweitert die Definition von Medizinprodukten gemäß MDR insbesondere um die Aspekte „Vorschläge zur Eindämmung”, „Informationen zur …” und „ein Hilfsmittel für …”. Somit umfasst diese Definition wesentlich mehr Produkte als die Definition „Medizinprodukt” gemäß MDR. So kann z. B. eine App, die Informationen von einem Patienten an einen Arzt für eine Diagnose liefert, nach dieser Definition als Medizinprodukt angesehen werden, obwohl die App selbst keine Diagnose erstellt.
Dies erweitert die Definition von Medizinprodukten gemäß MDR insbesondere um die Aspekte „Vorschläge zur Eindämmung”, „Informationen zur …” und „ein Hilfsmittel für …”. Somit umfasst diese Definition wesentlich mehr Produkte als die Definition „Medizinprodukt” gemäß MDR. So kann z. B. eine App, die Informationen von einem Patienten an einen Arzt für eine Diagnose liefert, nach dieser Definition als Medizinprodukt angesehen werden, obwohl die App selbst keine Diagnose erstellt.
1.1.3 Definition MDCG
Dennoch ist auch diese Definition nicht vollumfassend auf alle Produkte anwendbar und schafft keine klare Abgrenzung, wann ein Produkt kein Medizinprodukt ist. Daher werden weiterhin die Anforderungen der MDCG (Medical Device Coordination Group) MDCG 2019-11 Guidance on Qualification and Classification of Software in Regulation (EU) 2017/745 – Medical Device Regulation (MDR) and Regulation (EU) 2017/746 – IVDR von Oktober 2019 betrachtet. In diesem Dokument sind weitere Definitionen aufgeführt, die berücksichtigt werden sollen. So ist in MDCG-Kapitel 3.1 ”Introduction to qualification criteria" aufgeführt:
MDCG 2019-11
”It is important to clarify that not all software used within healthcare is qualified as a medical device. For example, ”Simple search", which refers to the retrieval of records by matching record metadata against record search criteria or to the retrieval of information does not qualify as medical device software (e. g. library functions)."
”It is important to clarify that not all software used within healthcare is qualified as a medical device. For example, ”Simple search", which refers to the retrieval of records by matching record metadata against record search criteria or to the retrieval of information does not qualify as medical device software (e. g. library functions)."
An dieser Stelle wird klar definiert, dass ein einfaches Durchsuchen von Aufzeichnungen oder Informationen ein Produkt nicht als medizinische Software qualifiziert. Als Beispiel wird hier eine Library Function angegeben:
”However, software which is intended to process, analyse, create or modify medical information may be qualified as a medical device software if the creation or modification of that information is governed by a medical intended purpose. For example, the software which alters the representation of data for a medical purpose would qualify as a medical device software. (e. g. ”searching image for findings that support a clinical hypothesis as to the diagnosis or evolution of therapy" or ”software which locally amplifies the contrast of the finding on an image display so that it serves as a decision support or suggests an action to be taken by the user"). However, altering the representation of data for embellishment/cosmetic or compatibility purposes does not readily qualify the software as medical device software."
Medizinischer Zweck
Sobald Software dazu bestimmt ist, medizinische Informationen zu verarbeiten, zu analysieren, zu erstellen oder zu modifizieren, ist diese Software als medizinische Software zu qualifizieren, wenn diese Erstellung oder Modifikation der Informationen einen medizinischen Zweck hat.
Sobald Software dazu bestimmt ist, medizinische Informationen zu verarbeiten, zu analysieren, zu erstellen oder zu modifizieren, ist diese Software als medizinische Software zu qualifizieren, wenn diese Erstellung oder Modifikation der Informationen einen medizinischen Zweck hat.
”Software intended for non-medical purposes (excluding MDR Annex XVI devices), such as invoicing or staff planning, does not qualify as a medical device software. These software do not fall under the Medical Devices Regulations."
Es wird in diesem Absatz weiterhin definiert, dass eine Software ohne einen medizinischen Zweck nicht als medizinische Software gilt und sie daher auch nicht unter die Anforderung der Medical Device Regulation (MDR) fällt.
”A task such as e-mailing, web or voice messaging, data parsing, word processing, and back-up is by itself not considered as having a medical purpose."
Im letzten Satz sind Definitionen aufgeführt, die man zusammenfassend als „Kommunikation und Back-up” bezeichnen könnte. Diese Definitionen fallen demnach nicht unter einen medizinischen Zweck.
Der Vollständigkeit halber wird hier noch der Hinweis auf das Manual On Borderline And Classification In The Community Regulatory Framework For Medical Devices gegeben. In diesem Dokument sind Beispiele mit Begründung und Entscheidungen zu Medizinprodukten aufgeführt. Weiterhin weisen wir darauf hin, dass der Fokus in diesem Beitrag auf dem EU-Markt liegt und es für andere Märkte weitere Anforderungen oder Definitionen geben kann.
Abschließend sollte jeder Inverkehrbringer auf Basis der Zweckbestimmung definiert haben, ob es sich bei dem Produkt um ein Medizinprodukt handelt oder nicht. Über diese Entscheidung sollten Aufzeichnungen geführt und die Entscheidungen auf Basis der in diesem Abschnitt referenzierten Vorgaben getroffen werden.
1.2 Medical Device Software (MDSW)
Gemäß MDCG 2019-11 wird als Medical Device Software (MDSW) oder Medizinsoftware (Bezeichnung gemäß DIN EN 82304-1 Anhang A Begründung) eine Software bezeichnet, die gemäß der MDR oder der IVDR (In Vitro Diagnostic Regulation) einen medizinischen Zweck erfüllt, unabhängig davon, ob die Software eigenständig ist oder ein Medizinprodukt steuert oder beeinflusst.
Eine MDSW kann gemäß dieser Definition in zwei Hauptkategorien unterteilt werden:
| • | Software in a Medical Device |
| • | Software as a Medical Device (SaMD) |
Durch diese Unterteilung können regulatorische Anforderungen und Klassifizierungen je nach Art der Medical Device Software präziser angewendet werden.
1.2.1 Software in a Medical Device
Die Software, die Bestandteil eines Medizinprodukts ist und dieses steuert oder beeinflusst und in Zusammenhang mit einem Medizinprodukt betrieben wird, wird als Medizinproduktesoftware (Bezeichnung gemäß DIN EN 82304-1 Anhang A Begründung) bezeichnet. Diese Software wird nicht als eigenständiges Medizinprodukt zugelassen und erhält daher auch keine eigenständige Klassifizierung gemäß Anhang VIII MDR und keine eigenständige technische Dokumentation.
Integraler Bestandteil des Produkts
Stattdessen wird sie als integraler Bestandteil des Produkts betrachtet. Ein Beispiel für eine solche Software ist die Steuerung eines MRT-Geräts. In diesem Fall unterliegt die Software den Regularien und Anforderungen, die für das gesamte Medizinprodukt gelten, und wird als Teil desselben behandelt.
Stattdessen wird sie als integraler Bestandteil des Produkts betrachtet. Ein Beispiel für eine solche Software ist die Steuerung eines MRT-Geräts. In diesem Fall unterliegt die Software den Regularien und Anforderungen, die für das gesamte Medizinprodukt gelten, und wird als Teil desselben behandelt.
1.2.2 Software as a Medical Device (SaMD)
Software as a Medical Device (auch „Software-Medizinprodukt”– Bezeichnung gemäß DIN EN 82304-1 Anhang A Begründung) ist eine eigenständige Software mit einem eigenständigen medizinischen Zweck. Für diese Software wird somit eine Klassifizierung gemäß der MDR durchgeführt und eine eigene technische Dokumentation erstellt. Ein Beispiel für eine solche Software ist eine App mit der eine psychiatrische Therapie durchgeführt wird wie z. B. tägliche Übungen bei Stress oder Burn-out-Patienten.
Dabei kann diese Software als App, online oder auch als Web-App in Verkehr gebracht werden. Eine Web-App ist eine Webapplikation, die dem Nutzer auf dem Smartphone das Look and Feel einer App gibt, aber nicht auf dem Smartphone installiert, sondern über den Browser aufgerufen wird. Im Beitrag Compliance by Design − Entwicklung medizinischer Apps (s. Kap. 04011) sind besondere Anforderungen in Bezug auf die Compliance von medizinischen Apps aufgeführt.
Besonderheit „Gesundheitssoftware”
In der DIN EN 82304-1 wird der Begriff „Gesundheitssoftware” verwendet. Dazu wird im Normanhang A „Begründung” folgende Definition aufgeführt:
In der DIN EN 82304-1 wird der Begriff „Gesundheitssoftware” verwendet. Dazu wird im Normanhang A „Begründung” folgende Definition aufgeführt:
„Das International Medical Device Regulators Forum (IMDRF) veröffentlichte das Dokument SaMD WG/N10FINAL, Software as a Medical Device (SaMD): Key Definitions [19]. Wenn Gesundheitssoftware einem medizinischen Zweck dient und nicht mit spezifischer Hardware betrieben werden soll, dann ist sie mit einer Software als Medizinprodukt identisch.”
2 Relevante Normen
In den ersten Abschnitten dieses Beitrags wurde erläutert, dass es sich bei Software um Medizinprodukte, nicht eigenständige Medizinprodukte oder auch Nicht-Medizinprodukte handeln kann. In den nachfolgenden Abschnitten liegt der Fokus auf der Entwicklung von Software as a Medical Device (SaMD).
Bei der Entwicklung von Medizinprodukten wird gemäß Artikel 8 Absatz 1 MDR die Anwendung harmonisierter Normen gefordert:
| Artikel 8 Absatz 1 MDR | |
| (1) | Bei Produkten, die harmonisierten Normen oder den betreffenden Teilen dieser Normen entsprechen, deren Fundstellen im Amtsblatt der Europäischen Union veröffentlicht worden sind, wird die Konformität mit den Anforderungen dieser Verordnung, die mit den betreffenden Normen oder Teilen davon übereinstimmen, angenommen. Unter Absatz 1 gilt auch für System- oder Prozessanforderungen, die gemäß dieser Verordnung von den Wirtschaftsakteuren oder Sponsoren einzuhalten sind, einschließlich der Anforderungen im Zusammenhang mit den Qualitätsmanagementsystemen, dem Risikomanagement, den Systemen zur Überwachung nach dem Inverkehrbringen, den klinischen Prüfungen, der klinischen Bewertung oder der klinischen Nachbeobachtung nach dem Inverkehrbringen. |
Prozessnormen
Es wird hier explizit gefordert, dass die Anwendung von harmonisierten Normen auch auf System- oder Prozessanforderungen angewendet werden muss. Ein Hersteller muss demnach mindestens die EN ISO 13485 und die EN ISO 14971 als „Prozessnormen” berücksichtigen, um einen Nachweis der Erfüllung der regulatorischen Anforderungen der MDR zu erbringen.
Es wird hier explizit gefordert, dass die Anwendung von harmonisierten Normen auch auf System- oder Prozessanforderungen angewendet werden muss. Ein Hersteller muss demnach mindestens die EN ISO 13485 und die EN ISO 14971 als „Prozessnormen” berücksichtigen, um einen Nachweis der Erfüllung der regulatorischen Anforderungen der MDR zu erbringen.
Besonders herausfordernd ist das Verständnis von mindestens fünf Systemnormen für einen Hersteller von „Software as a Medical Device – Medical Apps”. Im Grunde kann man davon ausgehen, dass es kaum einen anderen Medizinproduktehersteller (in der EU) gibt, der mehr normative Entwicklungsprozessanforderungen berücksichtigen muss als ein Hersteller von „Software as a Medical Device – Medical Apps”.
Wichtigste Normen
Erschwerend kommt hinzu, dass die Normen eine Vielzahl von Schnittstellen haben, die zwar klar definiert, aber sehr komplex miteinander verbunden sind. So sollte ein Hersteller von „Software as a Medical Device – Medical Apps” mindestens folgende Normen (jeweils die aktuell gültige Version) kennen und berücksichtigen:
Erschwerend kommt hinzu, dass die Normen eine Vielzahl von Schnittstellen haben, die zwar klar definiert, aber sehr komplex miteinander verbunden sind. So sollte ein Hersteller von „Software as a Medical Device – Medical Apps” mindestens folgende Normen (jeweils die aktuell gültige Version) kennen und berücksichtigen:
| 1. | EN ISO 13485 Medizinprodukte – Qualitätsmanagementsysteme – Anforderungen für regulatorische Zwecke |
| 2. | EN ISO 14971 Medizinprodukte – Anwendung des Risikomanagements auf Medizinprodukte |
| 3. | DIN EN 62366-1 Medizinprodukte – Teil 1: Anwendung der Gebrauchstauglichkeit auf Medizinprodukte |
| 4. | DIN EN 62304 Medizingeräte-Software – Software-Lebenszyklus-Prozesse |
| 5. | DIN EN 82304-1 Gesundheitssoftware – Teil 1: Allgemeine Anforderungen für die Produktsicherheit |
2.1 DIN EN 62304 und DIN EN 82304-1
Für die Entwicklung von Software as a Medical Device (SaMD) sollten besonders zwei Normen berücksichtigt werden:
| • | die DIN EN 62304 „Medizingeräte-Software – Software-Lebenszyklus-Prozesse” |
| • | und die DIN EN 82304-1 „Gesundheitssoftware – Teil 1: Allgemeine Anforderungen für die Produktsicherheit”. |
Die DIN EN 62304 war bereits unter der MDD harmonisiert, und man kann davon ausgehen, dass diese Norm auch unter der MDR harmonisiert wird.
Um die Anforderungen der beiden Normen zu verstehen, betrachten wir zunächst den Anwendungsbereich. Die DIN EN 62304 stellt einen Rahmen von Lebenszyklusprozessen mit Aktivitäten und Aufgaben bereit und gilt für die Entwicklung und Wartung von Medizinproduktesoftware. Dies trifft zu, wenn die Software selbst ein Medizinprodukt ist oder wenn sie ein eingebetteter oder integraler Bestandteil eines fertigen Medizinprodukts ist. Im Anwendungsbereich der DIN EN 62304 ist im Normkapitel 1.2 die wichtige Anmerkung 1 aufgeführt:
„Diese Norm kann benutzt werden für die Entwicklung und Wartung von Software, die ein eigenständiges Medizinprodukt ist. Zusätzliche Entwicklungsaktivitäten sind aber auf der Systemebene erforderlich, bevor diese Art von Software in Betrieb gehen kann. Diese Systemaktivitäten werden nicht durch diese Norm abgedeckt, sie finden sich in IEC 82304-1.” (DIN EN 62304 (VDE 0750-101):2016-10 Normkapitel 1.2 Anmerkung 1)
Die DIN EN 82304-1 behandelt den gesamten Lebenszyklus von „Gesundheitssoftwareprodukten”. Die DIN EN 82304-1 gilt explizit nicht für Gesundheitssoftware, die dafür vorgesehen ist, Teil einer spezifischen Hardware zu werden. Die Norm verweist im Normkapitel 2 auf die Anwendung der DIN EN 62304.
Es gibt also einen direkten Zusammenhang zwischen den zwei Normen. Diese Zusammenhänge sind im Anhang A2 der DIN EN 82304-1 grafisch dargestellt.

In Abbildung 1 ist dargestellt, wie die Anwendungsbereiche der beiden Normen zu verstehen sind und dass die gemeinsame Schnittmenge die „Nur-Software-Medizinprodukte” sind. Klassischerweise fallen unter diese Kategorie die „Software as a Medical Device – Medical Apps”.
2.2 Herausforderungen bei der Anwendung der DIN EN 62304 in der Praxis
Wie bereits im Abschnitt 2.1 erläutert, bezieht sich der Anwendungsbereich der DIN EN 62304 auf Software, die selbst ein Medizinprodukt oder ein eingebetteter oder integraler Bestandteil eines fertigen Medizinprodukts ist.
Da die DIN EN 62304 beide Produkte berücksichtigt, führt dies in der praktischen Umsetzung oftmals zu Herausforderungen. Für „Software as a Medical Device – Medical Apps” ist besonders die Spezifikation der Anforderungen nicht eindeutig definiert. Dies wird deutlich, wenn man das V-Modell gemäß DIN EN 62304 betrachtet.

Die DIN EN 62304 behandelt die Entwicklung der Software beginnend mit der Spezifikation der Softwareanforderungen und endend mit dem verifizierten Software Subsystem. Die Anforderungen an PEMS (Programmable Electrical Medical Systems) und PESS (Programmable Electrical Sub Systems) stammen aus der Normreihe 60601. Der Einstieg in die Normanforderungen ist daher mit dem dargestellten V-Modell aus der DIN EN 62304 schwer verständlich. Daraus resultiert, dass es keine Vorgaben für die Ermittlung der Anforderungen an das Produkt gibt. Für „Software as a Medical Device – Medical Apps” sind die Anforderungen an die Software gemäß DIN EN 62304 nur aus dem unteren Teil des V-Modells zu erfüllen. Diese Anforderungen beziehen sich ausschließlich auf Softwareanforderungen.
2.3 Schnittstellen der Normen
Die DIN EN 82304-1 hat eine eigene Prozessdarstellung im Normanhang A3 aufgeführt. Wie bereits in Abschnitt 2.1 beschrieben, verweist die DIN EN 82304-1 bei vielen technischen Prozessanforderungen auf die DIN EN 62304.
Die Übersicht aus Normanhang A3 (s. Abb. 3) stellt den Fokus der DIN EN 82304-1 dar. Hier wird die Schnittstelle zu den Anforderungen der DIN EN 62304 deutlich.

Die DIN EN 82304-1 stellt für „Software as a Medical Device – Medical Apps” genau die in der DIN EN 62304 fehlenden Anforderungen dar und legt diese fest. Diese Anforderungen sind in den Normkapiteln 4 bis 8 der DIN EN 82304-1 aufgeführt.
2.4 Idealisiertes Modell der kombinierten Anforderungen

Wenn man die Anforderungen der beiden Normen in einem V-Modell mit dem Fokus auf „Software as a Medical Device – Medical Apps” darstellen würde, könnte das Ergebnis eine Darstellung wie in Abbildung 4 sein. In dieser Darstellungsform sind die PEMS- und PESS-Anforderungen durch die Anforderungen der DIN EN 82304-1 ersetzt worden. Diese Verbindung der beiden Normen ist absolut sinnvoll, erfordert jedoch ein tiefes Verständnis für die Anforderungen beider Normen.
3 Entwicklungsanforderungen
Die Anforderungen an die Entwicklung von Software gemäß den Anforderungen der DIN EN 62304 ist bereits ausführlich in dem Beitrag Erfahrungen bei der Erstellung von Software als Medizinprodukt – Tipps zur Umsetzung der DIN EN 62304 (s. Kap. 07110) beschrieben.
Aus diesem Grund werden in den nachfolgenden Abschnitten die Anforderungen der DIN EN 82304-1 mit dem Fokus auf die Entwicklung von „Software as a Medical Device – Medical Apps” betrachtet.
Die Betrachtung orientiert sich am Gesundheitssoftware-Produktprozess (s. Abb. 3) und wird in drei Prozessbereiche unterteilt. Diese sind in den nachfolgenden Abschnitten aufgeführt.

3.1.1 Bedürfnisse der Anwender
Wie in jedem Entwicklungsprozess wird in der Übersicht des Gesundheitssoftware-Produktprozesses im Normanhang A.3 (s. Abb. 3) im ersten Prozessschritt die Ermittlung der Bedürfnisse der Anwender gefordert. Dieser Prozessschritt wird in der DIN EN 82304-1 nicht weiter erläutert. Es wird an dieser Stelle empfohlen, sich an den Vorgaben der DIN EN ISO 13485:2021-12 Normkapitel 7.3.3 „Entwicklungseingaben” zu orientieren und diese als Mindestanforderungen zu verstehen.
Weitere Anforderungen
Besonders zu erwähnen sind die Anforderungen, die in den nachfolgenden Prozessschritten nicht abgebildet werden. Dazu gehören:
Besonders zu erwähnen sind die Anforderungen, die in den nachfolgenden Prozessschritten nicht abgebildet werden. Dazu gehören:
| • | Anwendbare regulatorische Anforderungen (Als besonders relevante Anforderungen für Medical Apps sind z. B. Anforderungen aus der Verordnung über das Verfahren und die Anforderungen zur Prüfung der Erstattungsfähigkeit digitaler Gesundheitsanwendungen in der gesetzlichen Krankenversicherung (Digitale Gesundheitsanwendungen-Verordnung – DiGAV), Informationssicherheitsanforderungen oder Anforderungen an den Datenschutz zu nennen.) |
| • | soweit angemessen, Informationen, die aus früheren ähnlichen Designs abgeleitet wurden. (Diese Informationen können auch Informationen über IT-Sicherheitsstandards oder allgemein anerkannte Coding Rules sein.) |
3.1.2 Zweckbestimmung
Im ersten Schritt muss die Zweckbestimmung definiert werden. Dabei müssen immer das vorgesehene Anwenderprofil und die vorhergesehene Einsatzumgebung mitberücksichtigt und dokumentiert werden.
Initiale Risikobewertung
Es sollte eine initiale Risikobewertung durchgeführt werden, wobei die DIN EN 82304-1 betont, dass neben den Sicherheitsmerkmalen auch die Merkmale der Informationssicherheit, Konfigurationen und Schnittstellen zu anderen Produkten dokumentiert sein müssen. In Anmerkung 1 im Normkapitel 4.1 der DIN EN 82304-1 wird erwähnt, dass „kein formales vollständiges Risikomanagement wie z. B. nach ISO 14971 gefordert wird, die Durchführung der ersten Stufe dieses Prozesses jedoch als gute Praxis angesehen wird”. Für die in diesem Beitrag betrachteten Produkte „Software as a Medical Device – Medical Apps” sollten Hersteller immer die Anforderungen der ISO 14971 erfüllen. Daher wird empfohlen, unbedingt dem Risikomanagementprozess der ISO 14971 zu folgen.
Es sollte eine initiale Risikobewertung durchgeführt werden, wobei die DIN EN 82304-1 betont, dass neben den Sicherheitsmerkmalen auch die Merkmale der Informationssicherheit, Konfigurationen und Schnittstellen zu anderen Produkten dokumentiert sein müssen. In Anmerkung 1 im Normkapitel 4.1 der DIN EN 82304-1 wird erwähnt, dass „kein formales vollständiges Risikomanagement wie z. B. nach ISO 14971 gefordert wird, die Durchführung der ersten Stufe dieses Prozesses jedoch als gute Praxis angesehen wird”. Für die in diesem Beitrag betrachteten Produkte „Software as a Medical Device – Medical Apps” sollten Hersteller immer die Anforderungen der ISO 14971 erfüllen. Daher wird empfohlen, unbedingt dem Risikomanagementprozess der ISO 14971 zu folgen.
3.1.3 Nutzungsanforderungen
Die zu dokumentierenden Nutzungsanforderungen beginnen mit den Schnittstellenanforderungen, einschließlich der Anwenderschnittstellenanforderungen. Diese bilden im Verlauf der Nutzungsanforderungen die Grundlage für die Ableitung technischer Anforderungen. Zur strukturierten Umsetzung hat es sich als effektiv erwiesen, diesen Entwicklungsprozess durch eine systematische Nummerierung zu gliedern. Dadurch wird sichergestellt, dass die technischen Anforderungen auf den Nutzungsanforderungen basieren.
Um die Anwenderschnittstellenanforderungen angemessen zu berücksichtigen, wird auf die Prozessvorgaben der DIN EN 62366-1:2021-08 „Anwendung der Gebrauchstauglichkeit auf Medizinprodukte” verwiesen. Diese Norm ist ein wesentlicher Bestandteil bei der Entwicklung von Medizinprodukten.
Informationssicherheit
In den Nutzungsanforderungen müssen auch Anforderungen hinsichtlich der Unempfindlichkeit oder Anfälligkeit gegenüber anderer Software, die auf derselben Hardware betrieben wird, dokumentiert werden. Ebenso sind Anforderungen bezüglich Privatsphäre und Informationssicherheit zu erfassen. Dabei sollten vor allem die Grundsätze der Informationssicherheit, basierend auf der CIA-Triade (Confidentiality, Integrity und Availability), berücksichtigt werden. Zur Unterstützung wird auf die IEC/TR 80001-2-2 verwiesen, eine Richtlinie zur Offenlegung und Kommunikation von Sicherheitsanforderungen, Risiken und Kontrollen bei Medizinprodukten.
In den Nutzungsanforderungen müssen auch Anforderungen hinsichtlich der Unempfindlichkeit oder Anfälligkeit gegenüber anderer Software, die auf derselben Hardware betrieben wird, dokumentiert werden. Ebenso sind Anforderungen bezüglich Privatsphäre und Informationssicherheit zu erfassen. Dabei sollten vor allem die Grundsätze der Informationssicherheit, basierend auf der CIA-Triade (Confidentiality, Integrity und Availability), berücksichtigt werden. Zur Unterstützung wird auf die IEC/TR 80001-2-2 verwiesen, eine Richtlinie zur Offenlegung und Kommunikation von Sicherheitsanforderungen, Risiken und Kontrollen bei Medizinprodukten.
Begleitdokumentation
Weiterhin müssen in den Nutzungsanforderungen Anforderungen an die Begleitdokumentation wie beispielsweise die Gebrauchsanweisung aufgeführt werden. Die letzten beiden Anforderungen beziehen sich auf den Produktsupport sowie allgemeine Verweise auf Anforderungen, die sich aus geltenden Vorschriften ergeben, insbesondere hinsichtlich des Schutzes personenbezogener Daten. Dabei ist zu beachten, dass häufig auch nationale Vorschriften berücksichtigt werden müssen.
Weiterhin müssen in den Nutzungsanforderungen Anforderungen an die Begleitdokumentation wie beispielsweise die Gebrauchsanweisung aufgeführt werden. Die letzten beiden Anforderungen beziehen sich auf den Produktsupport sowie allgemeine Verweise auf Anforderungen, die sich aus geltenden Vorschriften ergeben, insbesondere hinsichtlich des Schutzes personenbezogener Daten. Dabei ist zu beachten, dass häufig auch nationale Vorschriften berücksichtigt werden müssen.
Abschließend wird in Normkapitel 4.3 der DIN EN 82304-1 die Verifizierung der Nutzungsanforderungen an das Gesundheitssoftwareprodukt gefordert. Der Hersteller muss in der Lage sein, diese Anforderungen zu erfüllen, und die Nutzungsanforderungen als Eingabe für die Systemanforderungen verwenden. Sollte es erforderlich sein, die Nutzungsanforderungen zu aktualisieren, muss der Hersteller sicherstellen, dass diese Aktualisierungen durchgeführt werden.
3.1.4 Systemanforderungen
Die Systemanforderungen für das Gesundheitssoftwareprodukt müssen klar festgelegt und dokumentiert werden. Es wird dringend empfohlen, eine eindeutige Nummerierung der Systemanforderungen beizubehalten, um eine klare Rückverfolgbarkeit zu den Nutzungsanforderungen, allgemeinen Anforderungen sowie Verifizierungs- und Validierungsmaßnahmen sicherzustellen.
Funktionalität für die Zweckbestimmung
Es ist wichtig, dass die Anforderungen die Funktionalität für die Zweckbestimmung umfassen. Die DIN EN 82304-1 enthält Mindestanforderungen, die bei der Festlegung der Anforderungen zu berücksichtigen sind, insbesondere im Hinblick auf „Software as a Medical Device – Medical Apps”. Diese Anforderungen umfassen Aspekte wie
Es ist wichtig, dass die Anforderungen die Funktionalität für die Zweckbestimmung umfassen. Die DIN EN 82304-1 enthält Mindestanforderungen, die bei der Festlegung der Anforderungen zu berücksichtigen sind, insbesondere im Hinblick auf „Software as a Medical Device – Medical Apps”. Diese Anforderungen umfassen Aspekte wie
| • | Interoperabilität (die Fähigkeit mit anderen Systemen zusammenzuarbeiten), | ||||||
| • | Lokalisierung, | ||||||
| • | Sprachunterstützung, | ||||||
| • | Spezifikationen für die Benutzerschnittstelle, einschließlich
|
Des Weiteren gelten viele der Anforderungen in der DIN EN 82304-1 im Allgemeinen auch für andere Medizinprodukte. Dazu gehören
| • | Maßnahmen aus dem Risikomanagement, |
| • | Anforderungen an die Software- oder Hardwareplattform sowie |
| • | Merkmale zur Erkennung und Reaktion auf Beeinträchtigungen der Informationssicherheit. |
Softwaretools
Besondere Aufmerksamkeit sollte der Verwendung von Drittanbietersoftware, Softwaretools und Plug-ins gewidmet werden, da diese erhebliche Risiken im Hinblick auf Informationssicherheit und Datenschutz mit sich bringen. Bei der Entwicklung einer „Software as a Medical Device – Medical Apps” müssen diese Risiken sorgfältig berücksichtigt werden. Es sollte unbedingt darauf geachtet werden, dass bei der Anwendung solcher Tools die Fähigkeit der Anbieter sichergestellt ist. Dies wird in der Regel durch die Auswahl von zertifizierten Anbietern sichergestellt. Davon unberührt ist es aber Aufgabe der Hersteller des Produkts, die Anforderungen an solche Tools ebenfalls zu spezifizieren.
Besondere Aufmerksamkeit sollte der Verwendung von Drittanbietersoftware, Softwaretools und Plug-ins gewidmet werden, da diese erhebliche Risiken im Hinblick auf Informationssicherheit und Datenschutz mit sich bringen. Bei der Entwicklung einer „Software as a Medical Device – Medical Apps” müssen diese Risiken sorgfältig berücksichtigt werden. Es sollte unbedingt darauf geachtet werden, dass bei der Anwendung solcher Tools die Fähigkeit der Anbieter sichergestellt ist. Dies wird in der Regel durch die Auswahl von zertifizierten Anbietern sichergestellt. Davon unberührt ist es aber Aufgabe der Hersteller des Produkts, die Anforderungen an solche Tools ebenfalls zu spezifizieren.
Eindeutig und prüfbar
Die festgelegten Systemanforderungen müssen vom Hersteller dahingehend geprüft werden, dass diese einander nicht widersprechen und absolut eindeutig formuliert sind. Weiterhin müssen die Anforderungen anhand von festgelegten Prüfkriterien prüfbar sein, damit die Umsetzung der Anforderungen verifiziert oder validiert werden kann. Bei der Verifizierung der Systemanforderungen muss der Hersteller sicherstellen, dass die Anforderungen identifizierbar sind. Zu jenem Zweck wurde in diesem Beitrag mehrfach erwähnt, dass es ein eindeutiges Nummerierungssystem für alle Anforderungen geben sollte. Wie bei den Nutzungsanforderungen muss der Hersteller auch bei den Systemanforderungen sicherstellen, dass diese bei Bedarf aktualisiert werden.
Die festgelegten Systemanforderungen müssen vom Hersteller dahingehend geprüft werden, dass diese einander nicht widersprechen und absolut eindeutig formuliert sind. Weiterhin müssen die Anforderungen anhand von festgelegten Prüfkriterien prüfbar sein, damit die Umsetzung der Anforderungen verifiziert oder validiert werden kann. Bei der Verifizierung der Systemanforderungen muss der Hersteller sicherstellen, dass die Anforderungen identifizierbar sind. Zu jenem Zweck wurde in diesem Beitrag mehrfach erwähnt, dass es ein eindeutiges Nummerierungssystem für alle Anforderungen geben sollte. Wie bei den Nutzungsanforderungen muss der Hersteller auch bei den Systemanforderungen sicherstellen, dass diese bei Bedarf aktualisiert werden.
Wichtiger Verweis
Im Normkapitel 4.5 der DIN EN 82304-1 gibt es einen sehr wichtigen Verweis auf die DIN EN 62304. Dieser ist in der Anmerkung 4 aufgeführt und lautet wie folgt: „Es besteht nicht notwendigerweise ein Unterschied zwischen den Softwaresystem-Anforderungen gemäß DIN EN 62304 und den Systemanforderungen gemäß DIN EN 82304-1”. Das beschreibt in exakter Weise die in Abbildung 4 dargestellte Schnittstelle zwischen den zwei Normen im Entwicklungsprozess.
Im Normkapitel 4.5 der DIN EN 82304-1 gibt es einen sehr wichtigen Verweis auf die DIN EN 62304. Dieser ist in der Anmerkung 4 aufgeführt und lautet wie folgt: „Es besteht nicht notwendigerweise ein Unterschied zwischen den Softwaresystem-Anforderungen gemäß DIN EN 62304 und den Systemanforderungen gemäß DIN EN 82304-1”. Das beschreibt in exakter Weise die in Abbildung 4 dargestellte Schnittstelle zwischen den zwei Normen im Entwicklungsprozess.
In Normkapitel 5 der DIN EN 82304-1 wird auf die Erfüllung der Anforderungen der IEC 62304 4.2, 4.3, Normabschnitte 5 bis 9, hingewiesen.
Restrisiken
Im Normkapitel 5 wird weiterhin ein wichtiger Hinweis auf die Umsetzung der Anforderungen der ISO 14971 aufgeführt. Dabei gibt es eine sehr interessante Formulierung. Es wird anerkannt, dass der Hersteller nicht alle Prozessschritte, die in der ISO 14971 angegeben sind, für beteiligte Komponenten erfüllen kann. In diesem Fall muss der Hersteller die Restrisiken betrachten und Risikobeherrschungsmaßnahmen für diese Restrisiken implementieren. Dazu sind beispielsweise Komponenten wie proprietäre Komponenten, Subsysteme, die nicht aus dem Gesundheitsbereich stammen, und ältere Software aufgeführt.
Im Normkapitel 5 wird weiterhin ein wichtiger Hinweis auf die Umsetzung der Anforderungen der ISO 14971 aufgeführt. Dabei gibt es eine sehr interessante Formulierung. Es wird anerkannt, dass der Hersteller nicht alle Prozessschritte, die in der ISO 14971 angegeben sind, für beteiligte Komponenten erfüllen kann. In diesem Fall muss der Hersteller die Restrisiken betrachten und Risikobeherrschungsmaßnahmen für diese Restrisiken implementieren. Dazu sind beispielsweise Komponenten wie proprietäre Komponenten, Subsysteme, die nicht aus dem Gesundheitsbereich stammen, und ältere Software aufgeführt.
Da sich dieser Beitrag auf „Software as a Medical Device – Medical Apps” konzentriert, ist es unwahrscheinlich, dass ältere Software relevant ist. Es ist jedoch wichtig zu beachten, dass Drittanbietersoftware, Softwaretools und Plug-ins verwendet werden können. Für diese Tools sollten Restrisiken als Blackbox bewertet werden, und es sollten entsprechende Risikokontrollmaßnahmen abgeleitet werden.
3.1.5 Validierungsplan
Die Anforderungen an den Validierungsplan sind gemäß Normkapitel 6 der DIN EN 82304-1 festgelegt. Die Validierung des Produkts erfolgt anhand der definierten Nutzungsanforderungen. Zur vollständigen Abbildung der Validierung ist die Erstellung eines Validierungsplans erforderlich. Dieser Plan sollte den Anwendungsbereich der Validierung sowie die spezifischen Validierungsaktivitäten umfassen. Es wird empfohlen, das Nummerierungssystem so zu strukturieren, dass eine klare Rückverfolgbarkeit zu den Nutzungsanforderungen gewährleistet ist.
Validierungsteam unabhängig von Entwicklungsteam
Es wird gefordert, dass Validierungsverfahren, Eingabedaten und Annahmekriterien klar definiert sind und dass die Qualifikationen des Personals festgelegt werden. Besondere Beachtung verdient die Anforderung, dass das Validierungsteam angemessen unabhängig vom Entwicklungsteam agieren muss. Diese Anforderung stellt insbesondere für kleine Entwicklungsteams in der Praxis häufig eine Herausforderung dar.
Es wird gefordert, dass Validierungsverfahren, Eingabedaten und Annahmekriterien klar definiert sind und dass die Qualifikationen des Personals festgelegt werden. Besondere Beachtung verdient die Anforderung, dass das Validierungsteam angemessen unabhängig vom Entwicklungsteam agieren muss. Diese Anforderung stellt insbesondere für kleine Entwicklungsteams in der Praxis häufig eine Herausforderung dar.
Für Validierungsverfahren sind
| • | Inspektionen, |
| • | Analysen, |
| • | Analogien, |
| • | Nachweise, |
| • | Simulationen, |
| • | Peer-Reviews, |
| • | Prüfungen oder |
| • | Zertifizierungen |
gängige Möglichkeiten.
3.2 Ausgaben
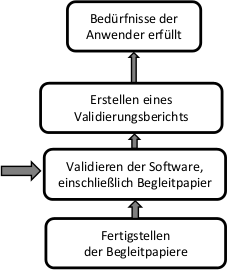
Die Anforderungen an die Ausgaben aus dem Gesundheitssoftware-Produktprozess beziehen sich im Wesentlichen auf die Identifizierung des Produkts und die Begleitpapiere.
Die Anforderungen an die Identifizierung des Produkts sind gemäß Normkapitel 7.1 der DIN EN 82304-1 definiert. Das Produkt muss mit einem Namen oder Handelsnamen des Herstellers sowie einem Produktnamen oder einer Typenbezeichnung und einer eindeutigen Versionskennzeichnung, wie beispielsweise einer Revisionsnummer oder einem Freigabedatum versehen sein. Für „Software as a Medical Device – Medical Apps” ist es zwingend erforderlich, dass eine UDI(Unique Device Identification)-Nummer vergeben wird.
Die Darstellung der Kennzeichnung auf der Startseite oder der Anmeldemaske wird in der Praxis als bewährte Vorgehensweise betrachtet.
3.2.1 Begleitpapiere
Der Hersteller muss Begleitpapiere zur Verfügung stellen, damit Anwender und Betreiber die Produkte wie vorgesehen installieren und betreiben können.
Die allgemeinen Anforderungen an die Begleitpapiere weichen nicht sonderlich von den Anforderungen für alle Medizinprodukte ab. Diese müssen verständlich sein, es muss eine Gebrauchsanweisung vorhanden sein, in der Einschränkungen und Anweisungen für den korrekten Betrieb und die korrekte Installation der Produkte enthalten sind. Für „Software as a Medical Device – Medical Apps” sind diese Anforderungen nahezu identisch mit den Anforderungen der MDR, die in jedem Fall von einem Medizinproduktehersteller umzusetzen sind.
Ein paar Anforderungen sind sehr spezifisch definiert, wie beispielsweise, dass alle Einstellmöglichkeiten zur Informationssicherheit für den Betrieb und die Nutzung des Produkts in der Gebrauchsanweisung aufgeführt sein müssen. Weiterhin sind ein paar spezifische Anforderungen an die Installation aufgeführt, die in der Gebrauchsanweisung beschrieben sein müssen.
Es wird an dieser Stelle empfohlen, die Anforderungen an die Begleitdokumentation gemäß Normkapitel 7 der DIN EN 82304-1 für die spezifische „Software as a Medical Device – Medical Apps” zu prüfen und die Erfüllung der Anforderungen sicherzustellen.
Das Normkapitel 7 der DIN EN 82340-1 enthält Anforderungen an Dokumenteninhalte und folgt damit nicht wie die anderen Normkapitel einem prozessorientierten Ansatz. Es kann wie eine Checkliste pro Produkt abgearbeitet werden.
3.2.2 Validierung
Die Durchführung der Validierung erfolgt gemäß den im Validierungsplan festgelegten Kriterien, einschließlich Umgebung, Zeitplan und Qualifikation des Personals. Sollten Abweichungen vom Validierungsplan erforderlich sein, sind diese im Validierungsbericht zu vermerken und zu bewerten.
Für den Fall, dass während der Validierung Anomalien festgestellt werden, müssen diese den in der DIN EN 62304 geforderten Problemlösungsprozess durchlaufen. In diesem Fall muss der betroffene Teil der Validierung im Anschluss wiederholt werden.
3.2.3 Validierungsbericht
Vom Validierungsteam ist ein Validierungsbericht zu erstellen, der über die durchgeführten Validierungstätigkeiten und die Validierungsergebnisse berichtet. Der Validierungsbericht muss die Validierungsbedingungen, die Ergebnisse sowie festgestellten Anomalien dokumentieren. Darüber hinaus sollte eine Zusammenfassung und eine Schlussfolgerung enthalten sein, die bescheinigen, dass das Produkt gemäß den Nutzungsanforderungen erfolgreich validiert wurde und für die beabsichtigte Verwendung geeignet ist.
3.3 Aktivitäten nach dem Inverkehrbringen

Die DIN EN 82304-1 behandelt den gesamten Lebenszyklus eines Produkts, einschließlich der Wartung der Software. Dabei soll sichergestellt werden, dass auch nach durchgeführten Wartungen die „Software as a Medical Device – Medical Apps” weiterhin die Anforderungen erfüllt und sicher betrieben werden kann.
Updates
In der Praxis werden die Wartungszyklen immer kürzer. Dies hängt mit den zunehmend agilen Entwicklungsprozessen und den Anforderungen an den aktuellen Stand der Technik zusammen. Besonders um die Informationssicherheit zu gewährleisten, ist es unabdingbar, regelmäßige Wartungen in Form von Updates durchzuführen. Nur so können Sicherheitslücken rechtzeitig geschlossen und die Funktionalität der Software sichergestellt werden.
In der Praxis werden die Wartungszyklen immer kürzer. Dies hängt mit den zunehmend agilen Entwicklungsprozessen und den Anforderungen an den aktuellen Stand der Technik zusammen. Besonders um die Informationssicherheit zu gewährleisten, ist es unabdingbar, regelmäßige Wartungen in Form von Updates durchzuführen. Nur so können Sicherheitslücken rechtzeitig geschlossen und die Funktionalität der Software sichergestellt werden.
Besonders diese Prozessschritte sind für Hersteller von „Software as a Medical Device – Medical Apps” schwer mit den regulatorischen Anforderungen zu vereinbaren. Für Hersteller ist dabei insbesondere die Anforderung an die Revalidierung wichtig und entscheidend.
Revalidierung
Der Hersteller muss sicherstellen, dass eine Revalidierung der durch die Wartung betroffenen Teile des Produkts stattfindet. Dazu muss der Validierungsplan angepasst werden, und der Hersteller muss sicherstellen, dass die Funktionalität mit jeder als unterstützt angegebenen Hardware- und Softwareplattform gewährleistet ist.
Der Hersteller muss sicherstellen, dass eine Revalidierung der durch die Wartung betroffenen Teile des Produkts stattfindet. Dazu muss der Validierungsplan angepasst werden, und der Hersteller muss sicherstellen, dass die Funktionalität mit jeder als unterstützt angegebenen Hardware- und Softwareplattform gewährleistet ist.
In der Praxis ist es äußerst empfehlenswert, das Produkt in unterschiedliche Softwarekomponenten bzw. Softwarearchitekturen zu unterteilen, auch wenn das nicht für alle Softwaresicherheitsklassen gefordert ist. Softwarekomponenten sollten in Bezug auf das Risiko und die Relevanz für die medizinischen Funktionen des Produkts eigenständig bewertet werden. Dadurch kann der Anteil an zu revalidierenden Softwarekomponenten minimiert und der Revalidierungsumfang reduziert werden, ohne die Sicherheit der Patienten, Anwender oder Dritter zu gefährden. Diese Unterteilung der Software in Komponenten ist eine elementare Anforderung der DIN EN 62304 und sollte daher ohnehin während der Softwareentwicklung durchgeführt werden.
4 Handlungsleitfaden für Hersteller
Nachfolgend wird ein Handlungsleitfaden zur Umsetzung von regulatorischen Anforderungen beschrieben. Dieser ist als Minimalanforderung zu verstehen und es wird explizit darauf hingewiesen, dass keine Haftung für die Vollständigkeit übernommen wird. Es wird empfohlen, diesen Leitfaden entsprechend den spezifischen Anforderungen und Gegebenheiten des Produkts und des Herstellers anzupassen.
| 1. | Der Hersteller muss entscheiden, ob es sich bei dem Produkt um ein Medizinprodukt handelt (s. Abschn. 1.1). | ||||||
| 2. | Der Hersteller muss die Zweckbestimmung definieren. | ||||||
| 3. | Der Hersteller muss entscheiden, ob es sich um eine Software in a Medical Device oder eine Software as a Medical Device – Medical App handelt (s. Abschn. 1.2). | ||||||
| 4. | Der Hersteller muss ein Qualitätsmanagementsystem aufsetzen und implementieren, dass die Anforderungen der ISO 13485 erfüllt.
| ||||||
| 5. | Der Hersteller muss eine Risikomanagementprozess gemäß den Anforderungen der ISO 14971 implementieren. | ||||||
| 6. | Der Hersteller muss einen Gebrauchstauglichkeitsprozess gemäß den Anforderungen der EN 62366 implementieren.Bis hierhin sind alle eingeführten Prozessanforderungen für jeden Hersteller von Medizinprodukten umzusetzen. Diese Anforderungen stellen das Fundament dar, auf dem die nachfolgenden Prozesse aufgesetzt werden können. | ||||||
| 7. | Der Hersteller muss den Gesundheitssoftwareprozess gemäß EN 82304 implementieren. Dabei sollte das Verständnis der Zusammenhänge der EN 82304 und der EN 62304 verstanden werden und die Schnittstellen klar definiert sein. Dazu kann das in diesem Beitrag in Abbildung 4 dargestellte idealisierte Schaubild hilfreich sein. | ||||||
| 8. | Der Hersteller muss den Softwareentwicklungsprozess gemäß EN 62304 implementieren. | ||||||
| 9. | Im letzten Schritt sollten alle weiteren geforderten Prozesse der EN 62304 implementiert werden. |
